Introduction
La fibrose pulmonaire est une maladie dans laquelle il y a une formation pathologique de tissu cicatriciel dans les poumons, de sorte qu’ils ne peuvent pas remplir pleinement leur fonction d’approvisionnement en oxygène de l’organisme. Cet état détériore considérablement la qualité de vie du patient. Dans certains cas, l’évolution de la maladie peut entraîner une insuffisance pulmonaire et/ou cardiaque et la mort. Les médicaments modernes ne peuvent pas inverser les changements pathologiques de la fibrose pulmonaire. D’autres méthodes de traitement de la pathologie peuvent inclure les cellules souches, puisque leurs propriétés étudiées impliquent la capacité à arrêter le processus inflammatoire, ainsi qu’à stimuler la récupération des tissus endommagés.
À propos de la fibrose pulmonaire
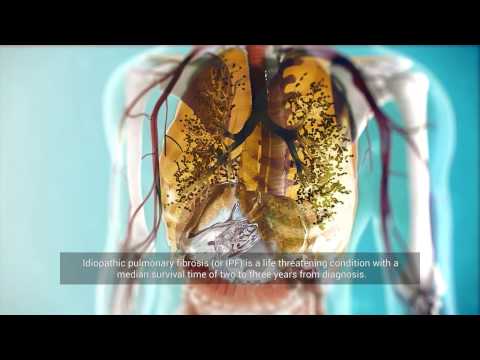
La fibrose pulmonaire est associée à la cicatrisation du tissu pulmonaire en raison de sa dégénérescence fibreuse. La croissance du tissu cicatriciel, causée en général par l’inflammation, entraîne une compression des alvéoles, qui ne peuvent plus remplir pleinement leur fonction, qui est d’assurer l’échange gazeux dans l’organisme. Les alvéoles sont les vésicules pulmonaires dans lesquelles l’air inhalé entre en contact avec le sang. Les lésions fibrotiques peuvent affecter une partie du poumon ou l’organe entier, dans un ou deux poumons.

La fibrose pulmonaire, ou pneumofibrose, est une maladie évolutive avec des modifications irréversibles du tissu pulmonaire, dont les complications et les conséquences incluent :
- Une insuffisance respiratoire chronique (manque d’oxygène dans l’organisme).
- L’hypertension pulmonaire (pression artérielle élevée dans les artères pulmonaires).
- Maladies pulmonaires cardiaques chroniques et autres maladies cardiovasculaires.
- Le cancer du poumon.
- Infection respiratoire secondaire (y compris le développement d’une pneumonie avec un risque élevé de décès).
Les causes de la fibrose pulmonaire
Il n’est pas toujours possible de déterminer la raison exacte qui provoque la cicatrisation des poumons. La croissance pathologique du tissu conjonctif dans les poumons peut être provoquée par des allergies, des infections pulmonaires, des facteurs génétiques, des radiations, des traumatismes et d’autres processus et circonstances.
Lorsqu’il est impossible de déterminer la cause exacte de la pathologie, on parle de fibrose pulmonaire idiopathique (FPI). Dans une telle maladie, une combinaison d’exposition environnementale, de facteurs génétiques et d’autres facteurs non identifiés est considérée comme un provocateur.
Il est également prouvé que le tabagisme (présent et passé), ainsi que des antécédents familiaux lourds (jusqu’à 20 % des patients), jouent un rôle. Lorsqu’au moins deux ou plusieurs membres de la famille biologique primaire (parent, frère, sœur ou enfant) ont eu des manifestations cliniques de la FIP, la maladie est classée comme une pneumonie interstitielle familiale (PIF). Les parents au premier degré des patients atteints de PIF ont un risque considérablement accru de développer une maladie pulmonaire interstitielle (MPI).
Fibrose pulmonaire interstitielle
Les maladies pulmonaires interstitielles (MPI), également connues sous le nom de maladie pulmonaire parenchymateuse diffuse (DPLD), sont un groupe de pathologies, dont la fibrose pulmonaire, dans lesquelles l’interstitiel (tissu conjonctif du poumon) est endommagé. Les DPLD sont associées à une inflammation et à une cicatrisation du tissu pulmonaire.
Le tissu interstitiel est le tissu conjonctif des poumons qui sert d’épine dorsale aux alvéoles et aux vaisseaux sanguins. Dans la fibrose, en raison de la croissance excessive du tissu conjonctif dans le poumon, les alvéoles sont comprimées et elles sont ensuite remplacées par du tissu conjonctif. En conséquence, l’échange gazeux dans les alvéoles devient difficile, ce qui entraîne une hypoxie — le corps commence à souffrir d’un manque d’oxygène.
Contrairement à la fibrose pulmonaire idiopathique (FPI), la cause de la MPI peut être déterminée de manière fiable. Ces raisons sont les suivantes :
- les lésions infectieuses;
- maladies auto-immunes / du tissu conjonctif;
- la prédisposition génétique;
- malignités;
- les substances nocives inhalées (vapeur de mercure, poussière minérale, etc.);
- certains médicaments (par exemple, certains antiarythmiques, la nitrofurantoïne et d’autres).
Contacter
Obtenez une consultation en ligne gratuite pour connaître les résultats attendus de la thérapie à base de cellules souches.

Medical Advisor, Swiss Medica doctor
La pandémie de coronavirus a augmenté le nombre de tous les cas de fibrose pulmonaire. On sait maintenant que la cicatrisation des poumons est l’une des conséquences les plus fréquentes et les plus graves de l’exposition au virus sur le corps humain. Le mécanisme de développement de la fibrose pulmonaire causée par COVID-19 est associé à une réponse immunitaire incorrecte, qui entraîne des lésions des cellules épithéliales pulmonaires et des cellules endothéliales microvasculaires, en particulier en combinaison avec la ventilation pulmonaire artificielle.

De plus, selon les chercheurs et les données statistiques, il est clair que les patients atteints d’une légère infection par COVID-19, même après une amélioration et leur sortie de l’hôpital, ont un risque potentiel caché de développer une fibrose pulmonaire.
Pour prévenir la progression de la fibrose pulmonaire, induite par le coronavirus, il est essentiel de commencer le traitement de cette complication le plus tôt possible.
Signes et symptômes de la fibrose pulmonaire
L’évolution clinique de la fibrose pulmonaire est associée à une diminution du volume des poumons, ce qui oblige l’organisme à s’adapter à cette conséquence négative. Les changements compensatoires se manifestent par un essoufflement, qui est le symptôme initial le plus fréquent signalé par les patients atteints de FPI. Au début, l’essoufflement ne se manifeste que pendant l’effort physique, mais plus tard — également au repos.
Le patient atteint de FPI est également confronté à des symptômes tels qu’une toux sèche, de la fatigue et une perte de poids inexpliquée.
En outre, des caractéristiques cliniques d’une maladie sous-jacente du tissu conjonctif (MTC), comme l’arthralgie (douleurs dans plusieurs articulations) ou des symptômes de sicca (sécheresse des glandes exocrines, en particulier des yeux et de la bouche), peuvent être observées.
Le principal signe externe de la FPI est connu sous le nom de «doigts gourdins», qui est une déformation des doigts ou des orteils caractérisée par un élargissement de l’extrémité des doigts. Ce symptôme est associé à un taux d’oxygène sanguin chroniquement bas.
Les premiers stades de la maladie se déroulent le plus souvent sans manifestation. Par exemple, chez les personnes asymptomatiques à risque de pneumonie interstitielle familiale (PIF), des signes de modifications histologiques dans le tissu pulmonaire peuvent être diagnostiqués.
Diagnostiquer la fibrose pulmonaire
Il existe une opinion selon laquelle la fibrose pulmonaire en tant que maladie est insuffisamment diagnostiquée. Le plus souvent, elle est détectée à un stade assez tardif. La maladie est souvent identifiée par une anomalie sur un scanner de la poitrine par tomographie à haute résolution (THR).
Il est prouvé que la radiographie du thorax est moins utile que la THR pour évaluer les patients suspectés de FPI.
La bronchoscopie avec lavage bronchoalvéolaire et la biopsie transbronchique sont également utilisées pour déterminer l’état du tissu pulmonaire.

De plus, les diagnostics de laboratoire de divers indicateurs (marqueurs biologiques) peuvent fournir des données objectivement mesurables sur les processus qui se déroulent dans l’organisme — tant normaux que pathologiques. Dans la fibrose pulmonaire, ces biomarqueurs aident à diagnostiquer la maladie, à révéler la sensibilité à la maladie et à prédire l’efficacité de certains médicaments. Ils peuvent également prédire avec précision l’évolution de la maladie et son pronostic chez un patient atteint de FPI.
Traitements traditionnels et modernes pour la MPI et la FPI
Bien que la fibrose pulmonaire puisse prendre des années pour se développer, la plupart des patients atteints de la maladie sont diagnostiqués à un stade avancé, alors que peu de choses peuvent être faites pour influencer sa progression. Les traitements existants visent à maintenir les performances pulmonaires du patient et un niveau de vie acceptable.
Dans la méthode courante de traitement de la pneumo fibrose, les hormones du cortex surrénal (prednisone) sont utilisées, et si elles ne sont pas assez efficaces, l’azathioprine et le cyclophosphane sont utilisés. On utilise également de l’oxygène pour l’inhalation. Récemment, il a été démontré que les médicaments pirfenidone et nintedanib ralentissent la progression de la FPI.
Cependant, la fibrose pulmonaire est une maladie évolutive et aucun agent ou traitement anti fibrotique (à l’exception d’une transplantation pulmonaire) n’est connu pour avoir un impact sur le faible taux de survie associé à cette maladie.
Les méthodes alternatives de traitement de la fibrose pulmonaire et de la maladie pulmonaire inflammatoire de l’intestin comprennent l’utilisation de produits à base de cellules souches. Les changements pathologiques qui se produisent dans les poumons en cas de fibrose sont considérés comme irréversibles, car l’organisme a une faible capacité de guérison du tissu cicatriciel. Cependant, lorsque nous introduisons des cellules souches dans l’organisme à une dose thérapeutique, cela suffit souvent à lancer les processus de régénération et à ralentir la cicatrisation du tissu pulmonaire. Ces cellules peuvent être injectées soit par voie intraveineuse (goutte à goutte), soit par inhalation pour une livraison ciblée des cellules à la zone endommagée.
Contacter
Contactez notre conseiller médical pour connaître les avantages que vous pouvez tirer de la thérapie à base de cellules souches.
Medical Advisor, Swiss Medica doctor
Thérapie à base de cellules souches pour la fibrose pulmonaire
La thérapie cellulaire est connue comme un traitement expérimental moderne qui utilise la capacité de l’organisme à se soigner lui-même.
Comment les cellules souches infusées agissent-elles dans la fibrose ? La particularité du ‘travail’ des cellules souches dans l’organisme est qu’elles répondent aux signaux envoyés par les cellules des tissus endommagés et produisent en réponse des cytokines (molécules protéiques qui transmettent les signaux intercellulaires) qui stimulent la formation des nouvelles cellules nécessaires.
Des études sur les cellules souches mésenchymateuses (CSM) allogéniques (de donneurs) ont montré qu’elles se fixent sur les sites de lésions pulmonaires et contribuent à la régénération des tissus, en diminuant l’inflammation chronique des voies respiratoires et en rétablissant l’équilibre du liquide alvéolaire dans les lésions pulmonaires aiguës.
Actuellement, d’autres essais cliniques sur les cellules souches pour les maladies pulmonaires sont en cours.
Quels sont les résultats possibles après une thérapie à base de cellules souches ?
En utilisant des cellules souches pour des perfusions intraveineuses dans le traitement de la fibrose pulmonaire, nous pouvons obtenir les effets suivants :
- Ralentir ou stopper le processus de cicatrisation en “calmant” le système immunitaire du patient (les cellules souches ont une capacité anti-inflammatoire).
- Régénération partielle du tissu pulmonaire.
- Formation de nouveaux réseaux vasculaires, qui contribuent au renouvellement des tissus.
- Soulagement des symptômes du patient.
Selon un essai clinique, les infusions endobronchiques de cellules souches ont augmenté le nombre d’indicateurs de la qualité de vie. 86 % des participants à l’étude, qui ont été observés pendant 12 mois, ont démontré un état fonctionnel et des performances physiques stables.
Ainsi, le rôle principal de la thérapie, basée sur l’introduction de cellules souches, est de ralentir la progression de la maladie, de stabiliser la fonction pulmonaire et de maintenir l’état actuel du patient.
Regardez la revue vidéo des patients atteints de BPCO (en italien) pour savoir comment la thérapie par cellules souches peut aider dans les maladies pulmonaires :

Une autre revue sur le traitement des cellules souches :

Sécurité des cellules souches dans la fibrose pulmonaire
Les essais cliniques de thérapie, basés sur des cellules souches adultes, ont démontré un profil de sécurité acceptable de cette approche. En particulier, il a été signalé que pendant toute la durée d’une étude utilisant 72 perfusions, aucun effet secondaire pouvant être considéré comme grave ou cliniquement significatif n’a été signalé.
La question de savoir si les CSM peuvent se différencier en fibroblastes, compte tenu de leur origine mésodermique commune, et accélérer la cascade fibreuse est actuellement débattue. Cependant, aucune étude préclinique ou humaine n’a démontré cette relation.
Aucun des patients n’a connu de formation anormale de tissus, comme l’a rapporté un scanner du corps entier effectué 12 mois après la première administration de cellules souches. Le même résultat positif de sécurité a été obtenu 24 mois après la première perfusion.
Une autre étude démontre que les perfusions de cellules souches ont été bien tolérées chez tous les patients et qu’aucun événement indésirable grave lié au traitement n’a été signalé.
Contacter
Contactez notre conseiller médical pour obtenir une consultation en ligne gratuite pour votre cas personnel >>>
Medical Advisor, Swiss Medica doctor
Liste des Références:
World Health Organisation. The top 10 causes of death. 24 May 2018.
Mesenchymal Stem Cell as Therapeutic Modality in Interstitial Pulmonary Fibrosis. Clinical Trial.


MD, Pediatrician, Regenerative Medicine Specialist